
Recently, Professor Qiu Zhengqing’s team from the Department of Pediatrics at PUMCH published their clinical research findings in the “World Journal of Pediatrics” (a tier 1 journal, or among the top 5%, as ranked by the Chinese Academy of Sciences). The study revealed a unique mutation spectrum of progressive pseudorheumatoid dysplasia in the Chinese population and made a preliminary exploration with the genotype-phenotype analysis. The research was supported by the National High Level Hospital Clinical Research Funding.

Progressive pseudorheumatoid dysplasia (PPRD) is a rare autosomal recessive disorder characterized by spondylo-epiphyseal dysplasia. The disease is caused by mutations in the CCN6 gene and is often misdiagnosed and mistreated. Due to its low incidence, media coverage is usually of individual cases or small case series and rarely of large case series in China.
To investigate the hotspot variants in Chinese PPRD patients, this study systematically analyzed the mutation spectrum and clinical phenotypic characteristics of Chinese PPRD patients and did genotype-phenotype correlation analysis. For this research, genetic analysis was conducted on suspected PPRD patients at PUMCH for confirmation. Cases were queried and their clinical data was systematically and retrospectively analyzed. International and domestic literature on PPRD in the Chinese population was reviewed, and cases without genetic testing results and cases that overlapped with those at PUMCH were deleted. Demographic information, gene mutations, clinical manifestations, and imaging data were collected for further analysis.
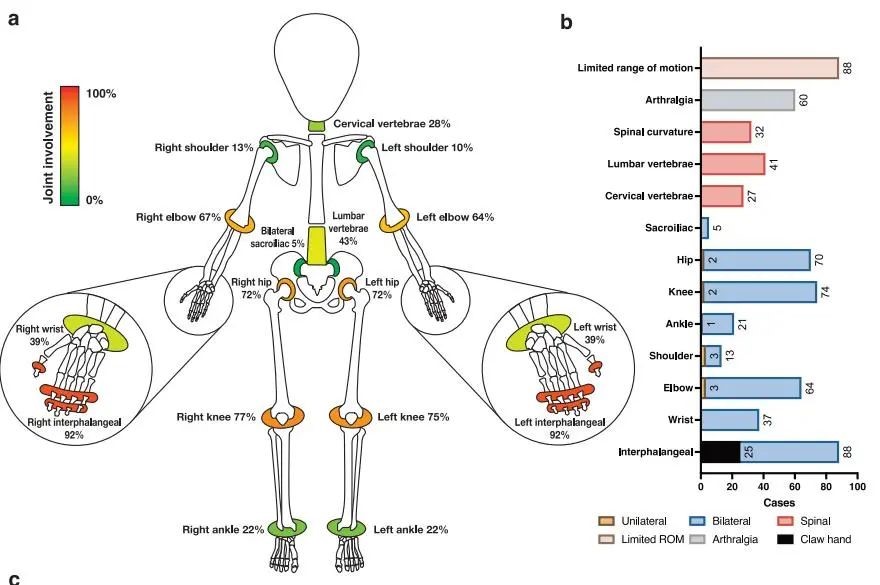
The study admitted a total of 105 PPRD patients (61 being PUMCH patients). Among them, 99 patients had detailed genetic information, and 96 patients had detailed clinical data. The main clinical manifestations of the Chinese PPRD patients include short stature, short neck, thoracic deformities, short trunk, polyarticular joint enlargement, flexion deformities and limited range of motion, spinal curvature, pronounced pelvic tilt in the standing position, and progressive gait changes ranging from normal gait to limping gait to waddling gait. In severe cases, patients were wheelchair-dependent and unable to walk at all or even developed secondary muscle atrophy. The median age of onset for the Chinese PPRD patients is 6 years old, with 72% of patients experiencing onset before the age of 7. Two patients had adult-onset PPRD, with onset occurring at the age of 24 and 29, respectively. The median age of diagnosis is 14 years old. Among pediatric patients, 70% had a height below the 3rd percentile (only 3 of 100 children are below this line), while among adult patients, 71% had a height below the 3rd percentile. There was no significant difference in the proportion of short stature between adult and pediatric patients. The affected sites, ranked by the incidence rate from high to low, are as follows: metacarpophalangeal joints, knee joints, hip joints, elbow joints, lumbar vertebrae, cervical vertebrae, ankle joints, shoulder joints, and sacroiliac joints (see the figure below).
In the 99 patients from 83 Chinese PPRD pedigrees, a total of 33 variants have been identified, including 9 novel variants. All variants are located in exons 2-5 of the CCN6 (NM_198239.2) gene, with no variant detected in exon 1. Among the 33 variants, only 7 have been reported in foreign patients, while 26 are exclusively found in Chinese population (see the figure below).
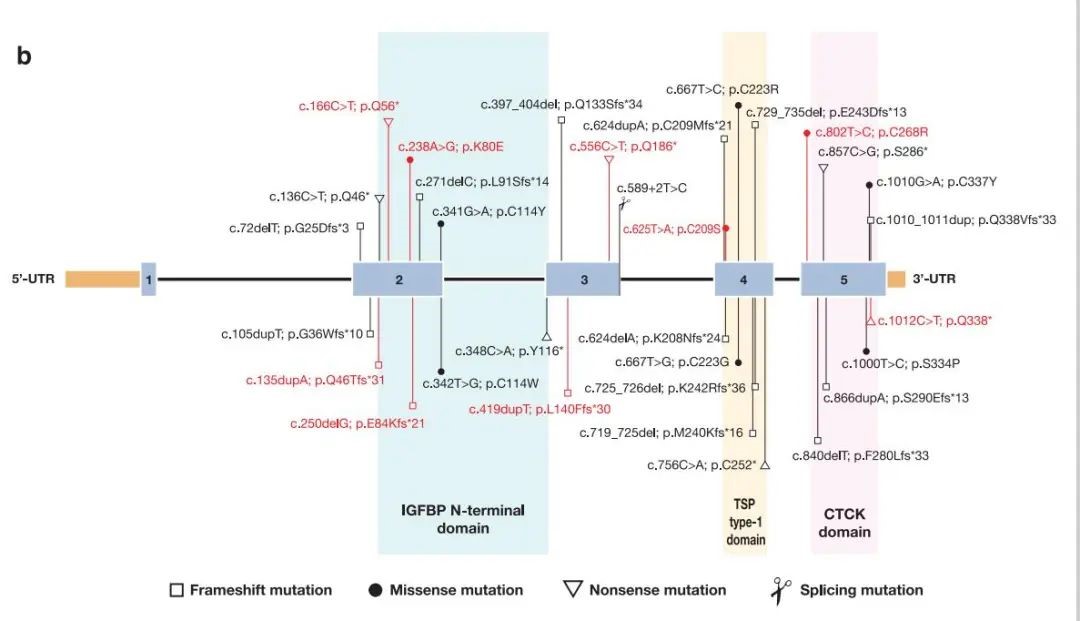
This research suggests that Chinese PPRD patients share a unique mutation spectrum. It also sheds some light on the genotype-phenotype relationship of the disease: among the five hotspot variants, c.624dupA is associated with later onset of disease, more extensive joint involvement, and a tendency to affect elbow joints; patients with only one variant identified may have a later onset. Compared with patients with two missense mutations, patients with two null mutations are affected in more joints, particularly in the cervical spine. The protein-truncating variant on exon 2 results in premature termination of translation into proteins, leading to complete loss of protein function, which may contribute to the shortening or deformity of long bones. The research findings can inform clinical diagnosis and treatment in a meaningful way.
Written by Wang Wei and Yan Xiaobo
Edited by Yan Xiaobo and Chen Xiao
Translated by Liu Haiyan
Reviewed by Li Yunwei and Wang Yao